应急审评审批下IVD产品信息汇总及历史进展分析
在突发公共卫生事件中,体外诊断(IVD)产品是疾病筛查、诊断和防控的关键工具。为应对紧急需求,各国药品监管部门通常会启动应急审评审批通道,以加速相关产品的上市进程。本文旨在汇总和分析在此类机制下获批的IVD产品关键信息及其历史发展脉络。
一、 应急审批机制概述
应急审评审批,是指在突发公共卫生事件(如新冠疫情、埃博拉疫情等)期间,监管机构为尽快将安全有效的诊断产品推向市场而采取的特殊审评程序。该程序在坚持科学、风险获益平衡和基本安全有效性的前提下,简化流程、缩短时限,例如接受更灵活的临床证据(如真实世界数据)、进行滚动审评或授予紧急使用授权(EUA)。中国的国家药品监督管理局(NMPA)、美国的食品药品监督管理局(FDA)以及欧盟的公告机构等均建立了相应的应急机制。
二、 主要IVD产品信息汇总(以新冠疫情为例)
在新冠疫情期间,全球范围内有大量IVD产品通过应急通道获批,主要包括:
- 核酸检测试剂:作为确诊的“金标准”,众多基于RT-PCR技术的试剂盒率先获得紧急授权,用于检测病毒核酸。
- 抗原检测试剂:以其快速(15-30分钟出结果)、便于基层使用的特点,在疫情中后期被广泛用于大规模筛查和居家自测。
- 抗体检测试剂:主要用于回顾性诊断和流行病学调查,检测人体感染后产生的IgM/IgG抗体。
- 其他技术平台:如基于CRISPR、等温扩增等新技术的检测产品也有部分获得授权。
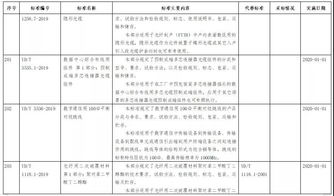
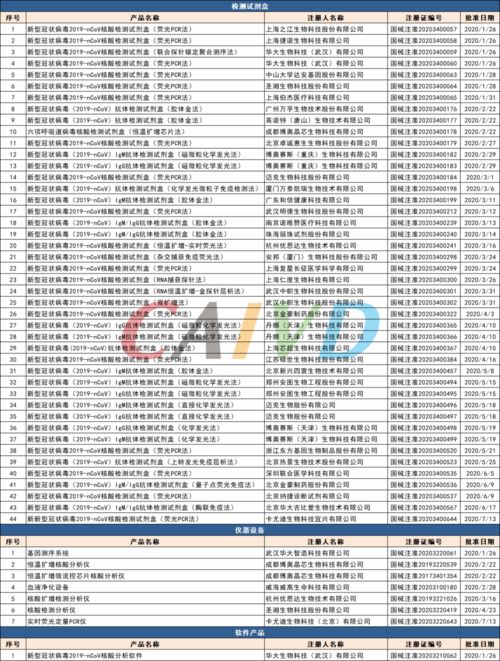
这些产品的审评信息通常可在监管机构官网的专题页面查询,关键信息包括产品名称、注册人/申请人、预期用途、批准日期、批准文号/授权编号以及重要的使用限制或警告说明。
三、 历史进展与关键节点分析
回顾近年来重大公共卫生事件中IVD产品的应急审批历程,可以观察到清晰的演进脉络:
- 响应速度显著提升:相较于早期的疫情(如2009年H1N1流感),新冠疫情期间监管机构建立应急通道、接受申请并作出首轮授权决策的速度明显加快,体现了机制和经验的成熟。
- 技术路径多元化:审批的产品从最初相对单一的PCR技术,迅速扩展到抗原、抗体检测,并包容了多种新兴技术,满足了不同场景下的诊断需求。
- 证据要求灵活而严谨:应急审批并非降低标准,而是更注重风险获益评估。监管机构逐步明确了应急状态下可接受的临床性能评价路径,如与已上市对照品的比对研究、有限的前瞻性临床研究等。
- 动态监管与退出机制:随着疫情变化和证据积累,监管机构会对已授权产品进行持续评估,对性能不佳或不再符合紧急需求的产品撤销授权,体现了全生命周期管理理念。
- 国际合作与标准协调:疫情中,WHO等国际组织发布紧急使用清单(EUL),各国监管机构间也加强了信息沟通与标准互认尝试,为全球协同应对奠定了基础。
四、 与展望
应急审评审批IVD产品的信息汇总和历史分析表明,建立高效、灵活且科学的应急监管机制至关重要。随着检测技术的不断革新和全球公共卫生体系的完善,应急审批机制有望进一步优化,例如:加强事前技术储备和平台建设;推动国际审评标准的进一步协同;利用真实世界证据支持应急决策;并完善从应急状态向常规注册的平滑过渡路径。持续跟踪和分析这些信息,不仅有助于行业把握研发与注册方向,也为应对未来可能的新发突发传染病提供了宝贵的经验借鉴。
如若转载,请注明出处:http://www.korings.com/product/2.html
更新时间:2026-04-18 12:41:16